Sequana: a set of NGS pipelines
This page serves as an additional set of resources for
the Sequana project. Sequana is comprised of a Python library, the
documentation of which is accessible on Read The Docs.
The source code for the Python library can be found on
GitHub.
If you use Sequana, please cite: Cokelaer et al.,
Journal of Open Source Software, 2017
(doi:10.21105/joss.00352).

One of the primary objective of the Sequana project is to offer a collection of NGS pipelines. The pipelines currently available are located on PyPI and the Sequana GitHub organisation page, each having their own repository. For instance, the RNA-seq pipeline can be found at sequana/rnaseq.
Pipelines
Sequana provides a collection of NGS pipelines. Each pipeline lives in its own repository on the Sequana GitHub organisation. Below are some highlighted pipelines:





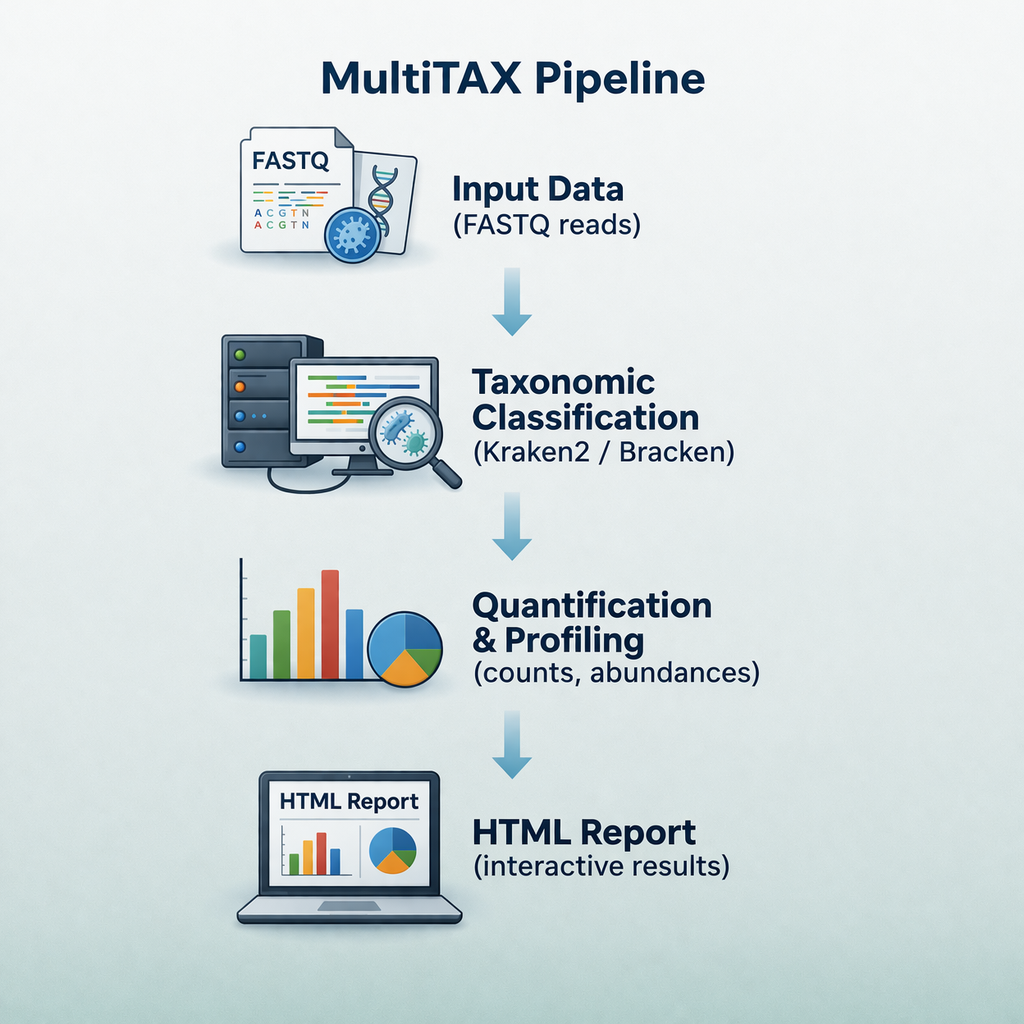

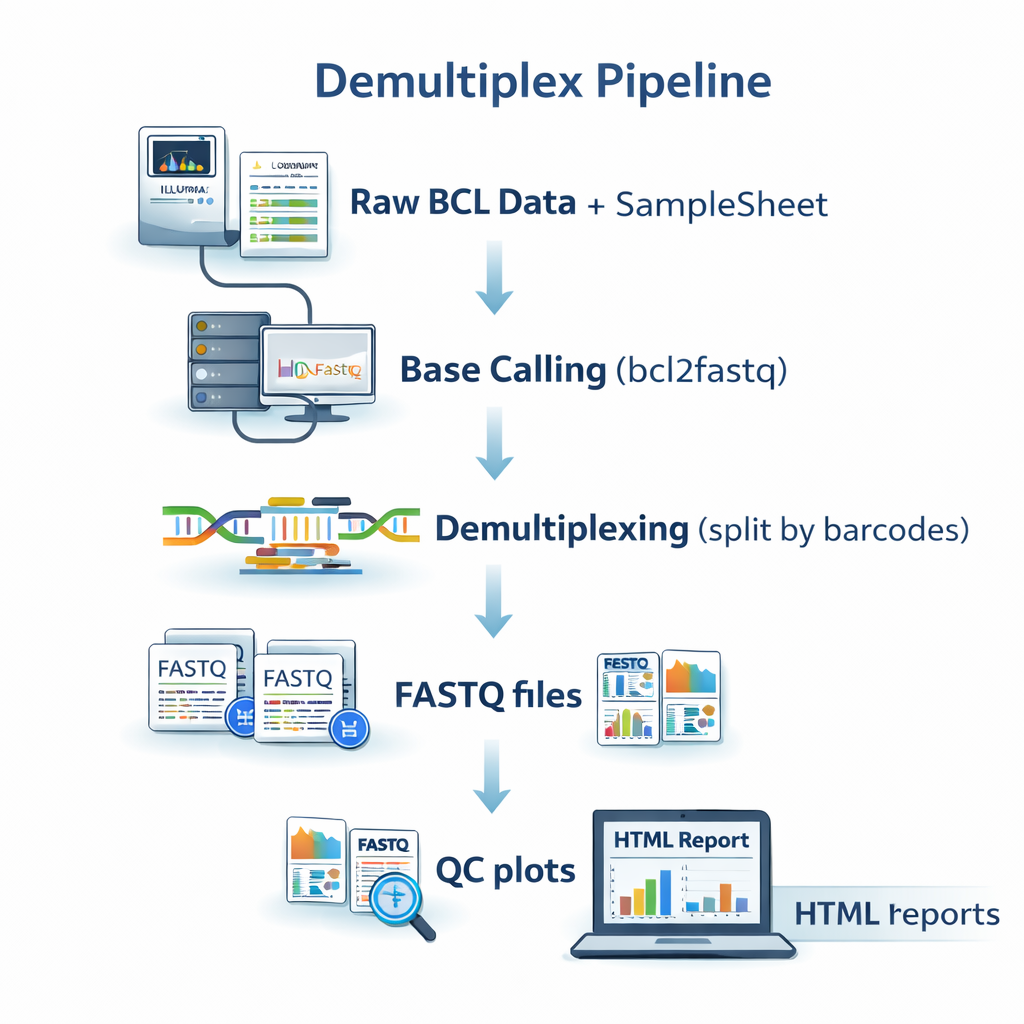

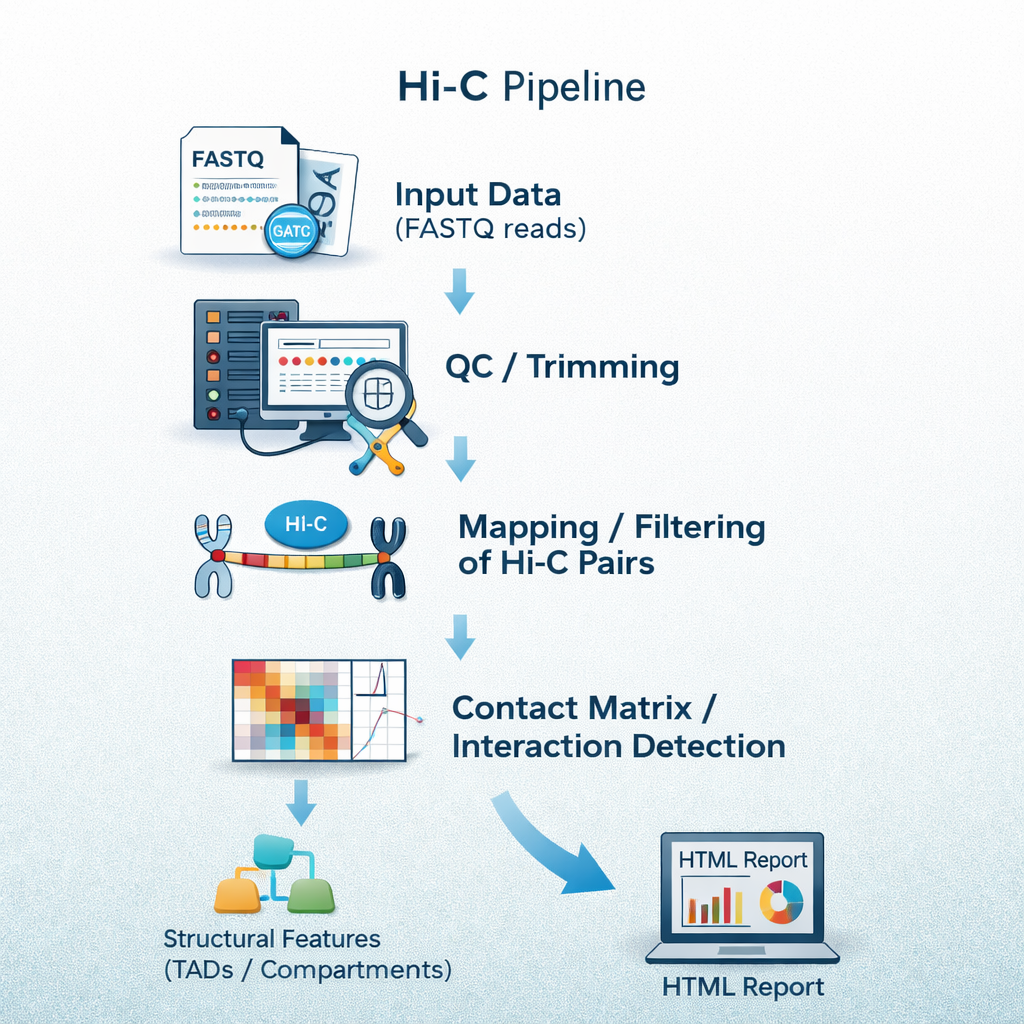



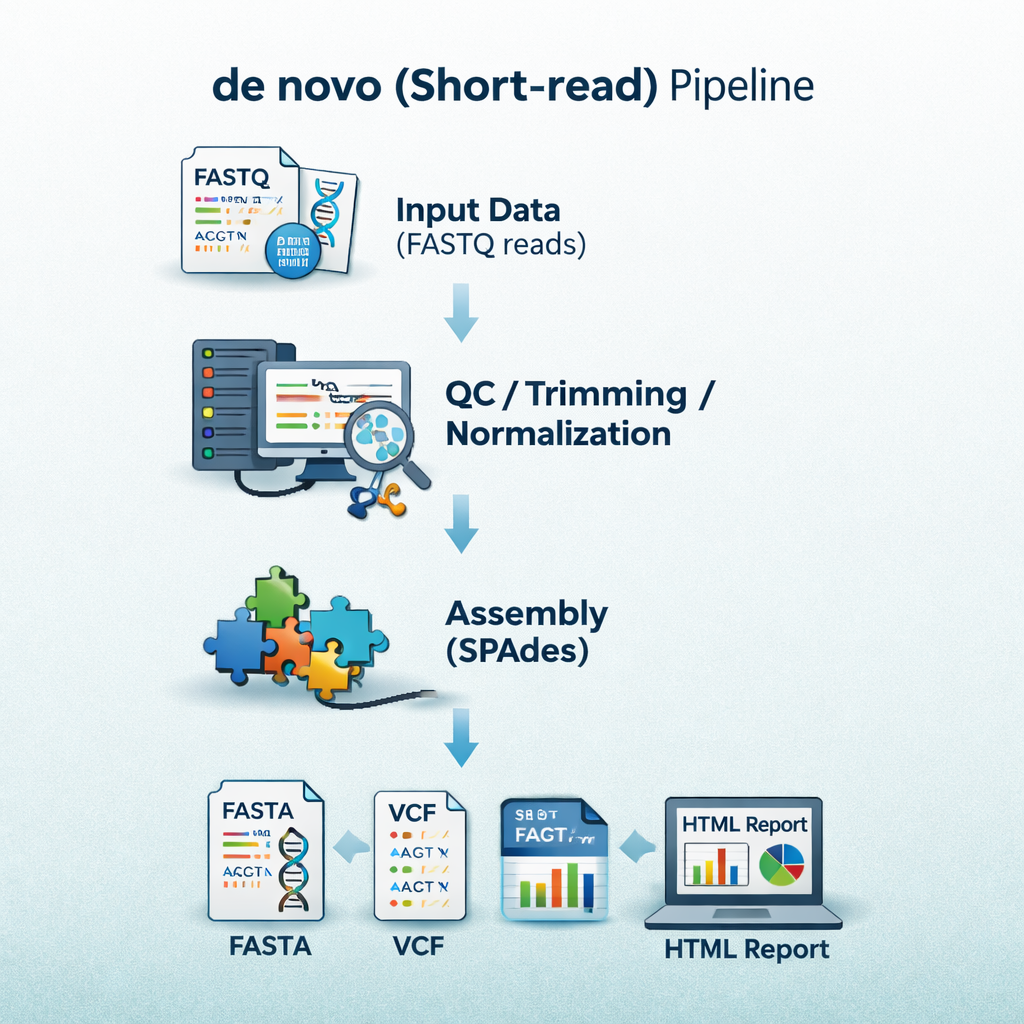



These are just a few highlights. Sequana encompasses many more pipelines — both public and private — covering a wide range of sequencing applications. A broader overview is available on github.com/sequana/sequana, the Sequana GitHub organisation, and the full documentation at sequana.readthedocs.io.
Tools & Standalone Applications
Sequana ships published tools and a set of standalone sub-commands invocable directly from
the command line (sequana <subcommand>), all distributed as part of the
sequana Python package.

sequana taxonomysequana summarysequana enrichmentsequana samplesheetsequana gtf_fixerTalks
Web Applications
Check your Illumina Sample Sheet
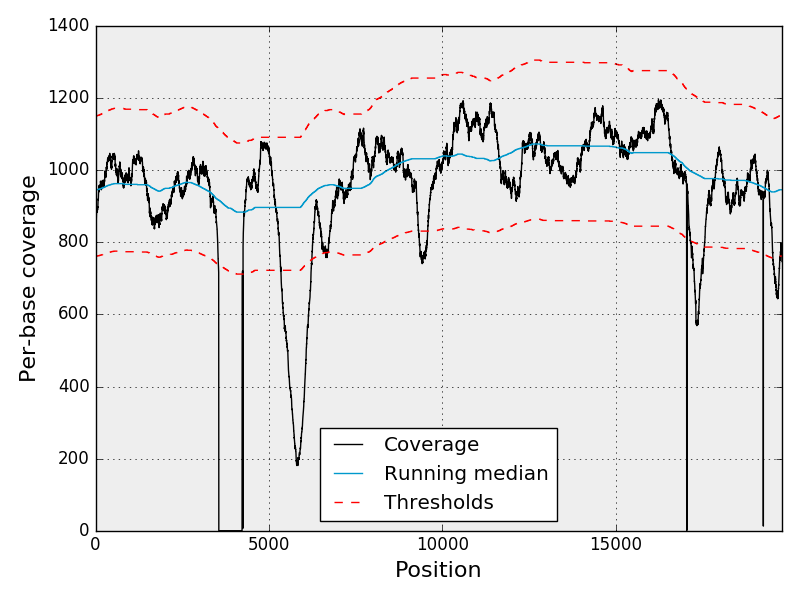
Report examples (click on the image)
The following was generated with the Sequana Coverage standalone application and keep here a it is reference in other places. This is the coverage along a virus genome where the black dot line represent the coverage, the blue is the median coverage, and red lines are the top and bottow thresholds for detecting event of interest.